Поиск сигналов
Задача 1 - Определение биологической роли транскрипционного фактора в бактерии
Наша задача - найти мотив связывания для транскрипционного фактора бактерии Staphylococcus aureus subsp. aureus N315. Для этого мы загрузили на сервер MEME 45 участков ДНК, с которыми он связывается.
Результатом выдачи является следующее лого:

Таблица с информацией о найденном мотиве
| Паттерн |
TTAADNNWHWDTTAA |
| Консенсус |
TTAATAAATAGTTAA |
| E-value |
3.1e-108 |
| Число сайтов |
45 |
| Длина |
15 нуклеотидов |
| PWM |
файл .txt с матрицей |
Teперь при помощи сервиса TOMTOM найдем мотив похожий на выбранный.
Для этого в html-выдаче МЕМЕ по ссылке Submit/Download -> Submit Motif -> Tomtom загрузим мотив в форму TOMTOM, после чего выберем в разделе Select target motifs параметры Prokaryote DNA -> Prokaryotes (RegTransBase v4), остальные будут заданы по умолчанию.
В итоге были найдены 4 похожие мотива. Ниже представлена лучшая находка и информация по ней.
.png)
Найденный MEME мотив изображён снизу, сверху найденный TOMMON мотив
| Название мотива |
ArcA_Gammaproteobacteria |
| Название ТФ |
Transcriptional dual regulator |
| Ссылка на ТФ |
Подробнее |
| E-value |
1.29e-02 |
| Длина |
14 нуклеотидов |
| PWM |
файл .txt с матрицей |
Эти мотивы не сильно различаются, однако в ArcA_Gammaproteobacteria после третьей позиции и в конце стоит С, что является главным отличием.
Теперь необходимо найти гены, регулируемые данным транскрипционным фатором. Для этого я использовал программу FIMO. Для этого я загрузил в программу найденный MEME и установил поиск в upstream region. Это оъясняется тем, что большинство транскрипционных факторов связываются с последовательностью до гена по направлению транскрипции, то есть как раз в upstream region.
Программа нашла 180 мотивов, всю информацию по ним можно посмотреть в этом файле.
Программа нашла множество мотивов, однако лишь несколько обладают p-value, достаточно маленьким, чтобы их можно было назвать достоверными.
Лучшие находки FIMO
| Название последовательности |
Координаты относительно гена |
Цепь |
p-value |
q-value |
Консенсус |
| ESK64900|ESK64900 |
42..57 |
+ |
7.31e-07 |
0.202 |
TTTAATTAAAATTTAA |
| ESK64841|ESK64841 |
91..106 |
- |
7.31e-07 |
0.202 |
TTTAATTAAAATTTAA |
| ESK64840|ESK64840 |
95..110 |
- |
4.59e-07 |
0.202 |
TTTAATAAATAGTTAG |
| ESK64840|ESK64840 |
189..219 |
+ |
4.59e-07 |
0.202 |
TTTAATAAATAGTTAG |
Первая находка представляет собой регулятор транскрипции, называемый AraC. Если отсутствует арабиноза, димер белка AraC подавляет структурный ген, связываясь с операторными участками araI1 и araO2, в результате чего ДНК образует петлю. Петля не даёт РНК-полимеразе связаться с промотором, тем самым блокируя транскрипцию. Когда в среде присутствует арабиноза, она связывает AraC и делает его неспособным связываться с операторами и репрессировать их. В результате происходит нарушение структуры петли ДНК. Таким образом, при присутствии арабинозы, AraC действует в качестве активатора.
Вторая находка является регулятором альдоза-1 эпимеразы. Эпимераза - фермент, который катализирует обратимые реакции превращения стереоизомеров, имеющих более одного асимметричного атома углерода. Больше я ничего не нашел про этот белок.
Третья и четвертая находки оказались белками с невыяснеными функциями.
Метилирование найденого сайта связывания транскрипционного фактора
Метилирование оснований в ДНК имеет место во всех пяти царствах живых существ, включая бактерий. Наиболее типичное метилированное основание, встречающееся повсеместно - C5-Метил-цитозин (m5C). Для бактерий, помимо этого, характерно наличие в ДНК N6 -метил-аденина (m6A) (есть у бактерий и низших эукариот) и N4-метил-цитозина (m4C) (исключительно бактериальный). Метильные группы m6A, m4C and m5C выступают из большой бороздки двойной спирали, которая является основным местом расположения сайтов связывания различных ДНК-связывающих белков. Поэтому метилирование A-C мотивов может в занчительной степени влиять на ДНК-белковые взаимодействия.
Формирование m6A, m4C и m5C катилизируется ДНК-метилтрансферазами, имеющимим собственные сайты узнавания. Подавляющая часть описанных к настоящему времени ДНК-метилтрансфераз являются участниками систем рестрикции-модификации, каждая из которых в типичном случае состоит из эндонуклеазы рестрикции и соответсвующей метилтрансферазы, "защищающей" необходимые участки от разрезания этой эндонуклеазой. Тем не менее существуют и одиночные метилтрансферазы, которые, вероятно, произшли в следствие утраты соответсвующих эндонуклеаз (наиболее известный пример - Dam-метилаза гамма-протеобактерий).
Если сайт связывания ДНК-метилазы пересекается с промоторным или регуляторным участком, метилирование может оказывать влияние на связывание транскрипционных факторов и, соответственно, модулировать транскрипцию. Метилирование участков, расположенных в относительной близости от сайта связвания транскрипционного фактора также может иметь подобный эффект[3].
С помощью программы fuzznuc пакета EMBOSS был проведен поиск сайтов метилирования, пересекающихся с найденными FIMO мотивами.
Для этого был взят файл с известными последовательностями сайтов метилирования (MT-sites) и файлы,
содержащие два лучших мотива и их окрестности в 60 нуклеотидов с каждой стороны (чтобы не упустить пересечения с сайтами, превышающими длину мотива) (mot1.fasta, mot2.fasta). Использованная команда:
fuzznuc -sequence mot1.fasta -pattern @mt-sites -outfile fuzznuc1.out
В результате работы программы нашлось достаточно много участков пересечения для исходного мотива (131),
однако примерно 60% находок в обоих случаях представляли собой 1 буквенные последовательности, которые,
на наш взгляд, не несут особого смысла. Если исключить их из числа значимых находок,
то фактическое число потенциальных участков пересечения становится гораздо меньше. Обобщение полученных данных представлено в Таблице 1.
| Ген |
Координаты мотива с окрестностью в 60 нуклеотидов |
Общая длина участка |
Количество находок |
Выходной файл |
| SA_RSO1910 |
391637..391772 |
135 |
131 |
fuzznuc1.out |
В целом пересечений последовательностей наших мотивов и сайтов связывания метилтрансфераз достаточно много. Тем не менее говорить однозначно о том, что метилирование
(если оно вообще действительно происходит в этих участках) как-то влияет на транскрипцию изучаемых генов, достаточно сложно.
Многие пересечения короткие и/или содержат протяженные участки, в которых нуклеотиды могут варьироваться (IUPAC Ambiguity Codes), поэтому они могли найтись случайно.
Некоторые пересечения могли попасть в окрестность и не затронуть сам мотив.
Самые длинные пересечения, не включающие N-позиций, имеют длину 6 нуклеотидов, более длинные пересечения, как правило, имеют посередине протяженный N-участок (например CCANNNNNNTTC - CCATTCATTTTC).
Достаточно часто встречаются повторяющиеся пересечения. Интересно, что по координатам они часто идут друг за другом.
Чтобы проверить есть ли в геноме нашей бактерии ДНК метилтрансферазы с той же специфичностью, что у тех, для которых были найдены пересечения с мотивами, геном Bacillus pumilus SAFR-032 был найден
в базе данных REBASE и была получена информация о системе рестрикции-модификации данной бактерии (Рис. 2). Имеющиеся метилтрансферазы обозначены фиолетовым, обе они являются метилтрансферазами первого и четвертого типа.
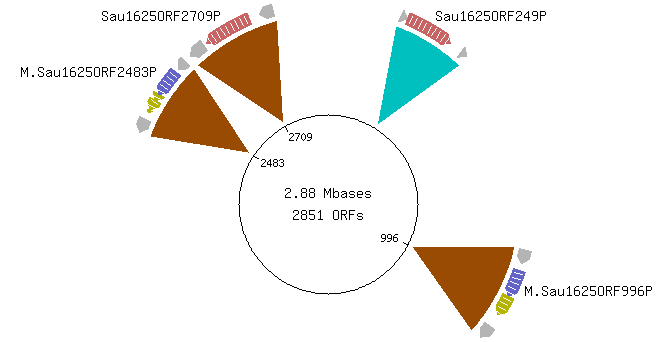

Рис. 2. Выдача REBASE. .
Вторая же, — M.Bpu32ORF656P (Рис. 4), — является цитозин-5 ДНК метилтрансферазой с предполагаемым участком узнавания GCWGC (W=A/C).
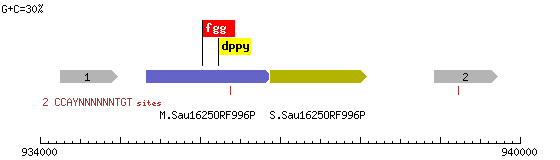
Рис. 4. Метилтрансфераза M.Sau16250RF996P и другие.
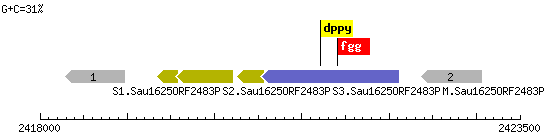
Рис. 5. Метилтрансфераза S1.Sau16250RF2483PS2 и другие.

Рис. 6. Метилтрансфераза Sau16250RF2709P и другие.
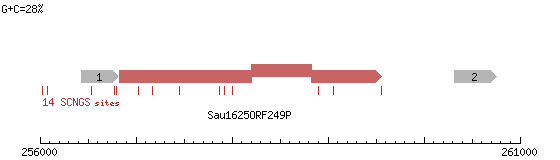
Рис. 7. Метилтрансфераза Sau16250RF249P и другие.
Для первого мотива данный сайт в найденных участках не присутствовал, однако были похожие сайты на 1 нуклеотид длиннее - GCATGC и GCAAGC. Для второго мотива самое похожее, что было найдено - GCATC, то есть mismatch в один нуклеотид. Так как точных совпадений найдено не было, вероятно, что метилирование не имеет серьезного влияния на связывание ТФ в нашем случае.
© Борисов Евгений/Цветков Роман 2017